
هدف از درمان این بیماری، کاهش مقدار فنیل آلانین در بدن به منظور پیشگیری از عقب ماندگی ذهنی کودک است.

به عقیده پژوهشگران، مشکلات و دردسرهای اقتصادی ناشی از ناهنجاریهای مادرزادی تنها بخشی از آسیبهایی است که این بیماریها به خانواده ها و جوامع وارد می کنند، در حالی که بخش مهم دیگر، مشکلات روانی و اجتماعی و عاطفی حاصل از آنهاست. در ضمن وجود این بیماران باعث می شود بخش مهمی از نیروهای بهداشتی و بودجه و اعتبارات درمانی جوامع صرف آنان شود و به دیگر بخشهای جامعه کمتر اختصاص یابد، به همین دلیل هرگونه تلاش برای کاستن از میزان این نقایص در نهایت به نفع کل جامعه خواهد بود.
ﻓﻨﻴﻞ ﻛﺘﻮﻧﻮری ﻳﻚ ﺧﻄﺎی ﻣﺘﺎﺑﻮﻟﻴﺴﻢ اﺳﺖ ﻛﻪ ﺑﻪ ﻋﻠﺖ ﻧﻘﺺ در آﻧﺰﻳﻢ ﻓﻨﻴﻞ اﻻﻧﻴﻦ ﻫﻴﺪروﻛﺴﻴﻼز ﻛﻪ ﻓﻨﻴﻞ اﻻﻧﻴﻦ را ﺑﻪ ﺗﻴﺮوزﻳﻦ ﺗﺒﺪﻳﻞ ﻣﻲ ﻛﻨﺪ اﻳﺠﺎد ﻣﻲ ﺷﻮد. ﻛﻤﺒﻮد اﻳﻦ آﻧﺰﻳﻢ ﻣﻨﺠﺮ ﺑﻪ اﻓﺰاﻳﺶ ﺳﻄﺢ ﻓﻨﻴﻞ اﻻﻧﻴﻦ ﺧﻮن ﻣﻲ ﺷﻮد. این بیماری از حدود ۴۰ سال پیش شناخته شده و امروزه یک بیماری کنترل شده است. تظاهرات بالینی بیمار تا ۶- ۵ ماهگی بسیار گمراه کننده است. متاسفانه تشخیص اغلب ، زمانی اتفاق می افتد که بیماری منجر به عقب ماندگی ذهنی کودک شده است و ضایعه مغزی به وجود آمده درمان ناپذیر شده است.بنابراین تنها و بهترین راه تشخیص این بیماری اندازه گیری غلظت خونی «فی» در بدو تولد نوزاد است. این آ زمایش باید هرچه زودتر در روزهای اول تولد انجام شود، زیرا تاخیر در تشخیص بیماری از هفته سوم به بعد خطرناک است و ممکن است صدمات مغزی به معلولیت دائم منتهی شود.
-
مقدمه:
PKUr که مخفف عبارت PHENYL KETONURIA فنیل کتونوری است، ازجمله بیماری های ژنتیکی است که به علت کمبود نوعی آنزیم در کبد نوزادان به وجود می آید. چون در ادرار مبتلایان ماده ای به نام فنیل کتون وجود دارد به همین دلیل بیماری را فنیل کتونوری نامیده اند.
ﻓﻨﻴﻞ ﻛﺘﻮﻧﻮری ﺷﺎﻳﻊ ﺗﺮﻳﻦ ﻓﺮم ﻳﻚ ﺑﻴﻤﺎری autosomal recessive ﺑﻨﺎم ﻫﻴﭙﺮ ﻓﻨﻴﻞ آﻻﻧﻴﻨﻨﻤﻴﺎﺳﺖ ﻛﻪ ﺑﻌﻠﺖ ﻛﻤﺒﻮد ﻳﺎ ﻋﺪم ﻓﻌﺎﻟﻴﺖ آﻧﺰﻳﻢ ﻓﻨﻴﻞ آﻻﻧﻴﻦ ﻫﻴﺪروﻛﺴﻴﻼز اﺳﻴﺪ در ﻛﺒﺪ، آﻣﻴﻨﻪ ﺿﺮوری ﻓﻨﻴﻞ آﻻﻧﻴﻦ ﺑﻪ ﺗﻴﺮوزﻳﻦ ﺗﺒﺪﻳﻞ ﻧﺸﺪه و ﺳﺒﺐ اﻓﺰاﻳﺶ ﻣﺰﻣﻦ ﻓﻨﻴﻞ آﻻﻧﻴﻦ ﺧﻮن و ﻏﻴﺮ ﭘﻴﺪاﻳﺶ ﻋﻮارﺿﻲ از ﺟﻤﻠﻪ آﺳﻴﺐ ﻣﻐﺰی ﻣﻲ ﻗﺎﺑﻞ ﺑﺮﮔﺸﺖ ﺷﻮد . ﻓﻨﻴﻞ ﻛﺘﻮﻧﻮری ﻳﻚ ﺧﻄﺎی ﻣﺘﺎﺑﻮﻟﻴﺴﻢ اﺳﺖ ﻛﻪ ﺑﻪ ﻋﻠﺖ ﻧﻘﺺ در آﻧﺰﻳﻢ ﻓﻨﻴﻞ اﻻﻧﻴﻦ ﻫﻴﺪروﻛﺴﻴﻼز ﻛﻪ ﻓﻨﻴﻞ اﻻﻧﻴﻦ را ﺑﻪ ﺗﻴﺮوزﻳﻦ ﺗﺒﺪﻳﻞ ﻣﻲ ﻛﻨﺪ اﻳﺠﺎد ﻣﻲ ﺷﻮد. ﻛﻤﺒﻮد اﻳﻦ آﻧﺰﻳﻢ ﻣﻨﺠﺮ ﺑﻪ اﻓﺰاﻳﺶ ﺳﻄﺢ ﻓﻨﻴﻞ اﻻﻧﻴﻦ ﺧﻮن ﻣﻲ ﺷﻮد. [1]
اختلال اصلی در این بیماری، تجمع اسید آمینه فنیل آلانین در مایعات بدن و سیستم عصبی است. تجمع این اسید آمینه به دلیل عدم وجود آنزیم مورد نیاز برای تبدیل فنیل آلانین به تیروزین رخ میدهد. تجمع غیرطبیعی این اسید آمینه در بدن کودک، خطرناک است و منجر به بروز اختلالاتی در مغز و پوست میشود. این بیماری از طریق ژنهای مغلوب وکروموزوم های معمولی از والدین به کودک منتقل می شود وموجب بروزعقب ماندگی نسبتاً شدیدمی گردد. دراین بیماری اسیدآمینه فنیل آلانین که درموادچربی زیاد یافت می شود دچارعدم سوخت وسازشده و مقدارآن درخون افزایش می یابد. نوزاد مبتلا به علت کمبود آنزیم مخصوصی در کبد خود، قادر به هضم فنیل آلانین نیست. ﺑﺪون درﻣﺎن اﻏﻠﺐ ﺑﻴﻤﺎران دﭼﺎر ﻋﻘﺐ اﻓﺘﺎدﮔﻲ ذﻫﻨﻲ ﻣﻲ ﺷﻮﻧﺪ. ﺑﻪ ﺧﻮﺑﻲ ﻣﺸﺨﺺ ﺷﺪه اﺳﺖ ﻛﻪ درﻣﺎن ﻣﻨﺎﺳﺐ ﻧﻮزادان در ﺧﻼل دو ﻫﻔﺘﻪ اول زﻧﺪﮔﻲ ﭘﻴﺎﻣﺪﻫﺎی ﺑﻴﻤﺎری را ﺗﻐﻴﻴﺮ ﻣﻲ دﻫﺪ و ﻣﻨﺠﺮ ﺑﻪ رﺷﺪ و ﻧﻤﻮ ﻃﺒﻴﻌﻲ در ﻧﻮزاد ﻣﻲ ﺷﻮد .[5- 2]
ﺗﺸﺨﻴﺺ ﺳﺮﻳﻊ، ﺑﻪ اﻳﻦ ﻋﻠﺖ در اوﻟﻴﻦ روزﻫﺎی زﻧﺪﮔﻲ اﻗﺪام اﺳﺎﺳﻲ اﺳﺖ. ﻏﺮﺑﺎﻟﮕﺮی اﻳﻦ ﺑﻴﻤﺎری ﺗﻮﺳﻂ آزﻣﻮﻧﻬﺎی ﻏﺮﺑﺎﻟﮕﺮی ﻧﻮزادی ﻳﻚ اﻗﺪام ﺳﻼﻣﺖ اﺟﺘﻤﺎﻋﻲ اﺳﺖ ﻛﻪ در ﭘﻨﺠﺎه ﺳﺎل اﺧﻴﺮ اﻧﺠﺎم ﻣﻲ ﺷﻮد. [ 6]
در ﻃﻮل ﭼﻨﺪ ﺳﺎل اﺧﻴﺮ ﭼﻨﺪﻳﻦ ﻣﻄﺎﻟﻌﻪ ﺑﺮ ﺑﻴﻤﺎران ﻓﻨﻴﻞ ﻛﺘﻮﻧﻮری در اﻳﺮان اﻧﺠﺎم ﺷﺪه اﺳﺖ. ]12-7]
-
ﺗﺎرﻳﺨﭽﻪ
ﻓﻨﻴﻞ ﻛﺘﻮﻧﻮری اوﻟﻴﻦ ﺑﺎر در ﺳﺎل Asborn Folling ،1934 ﻓﻨﻴﻞ ﭘﻴﺮوﻳﻚ اﺳﻴﺪ را در ادرار ﻓﺮزﻧﺪان دﭼﺎرﻋﻘﺐ ﻣﺎﻧﺪﮔﻲ ذﻫﻨﻲ ﻳﻚ ﺧﺎﻧﻮاده ﺷﻨﺎﺳﺎﻳﻲ [13] و آﻧــﺮا imbecilitas phenylpyrouvica ﻧﺎﻣﻴــﺪ. ﻳﻜــﺴﺎل ﺑﻌــﺪ Lionel Penrose اﻳــﻦ وﺿــﻌﻴﺖ را (PKU) ﻓﻨﻴــﻞ ﻛﺘﻮﻧــﻮری ﻧﺎﻣﮕﺬاری ﻛﺮد [14]. اواﺋﻞ 1960 ﮔﺎﺗﺮی روش bacterial inhibition assay را ﺑﻪ ﻣﻨﻈﻮر اﻧﺪازه ﮔﻴﺮی ﺳﻄﻮح ﺧﻮﻧﻲ ﻓﻨﻴﻞ آﻻﻧـﻴﻦ ﻣﻌﺮﻓـﻲ [13] ﻛﺮد ﻛﻪ ﺗﺴﺖ ﮔﺎﺗﺮی ﻧﺎﻣﻴﺪه ﺷـﺪ [14]. اواﺳـﻂ 1960 ﻓﺮﻣـﻮﻻی ﻛـﻢ ﻓﻨﻴﻞ آﻻﻧﻴﻦ، ﺑﻪ ﺻﻮرت ﺗﺠـﺎری ﻋﺮﺿـﻪ ﺷـﺪ. اواﺧـﺮ 1980 ﻣـﺸﺨﺺ ﮔﺮدﻳﺪ، ژن ﻣﺴﺌﻮل ﻛﻤﺒﻮد آﻧـﺰﻳﻢ ﻓﻨﻴـﻞ آﻻﻧﻴﻦ ﻫﻴﺪروﻛـﺴﻴﻼز روی ﻛﺮوﻣــﻮزوم 12q22-q24.1 ﻗــﺮار دارد و در 1990 ﻣﻴــﺰان ﻓﻨﻴــﻞ آﻻﻧﻴﻦ )60ﺗﺎ 360 ﻣﻴﻜﺮوﻣﻮل در ﻟﻴﺘﺮ ( 1 ﺗﺎ ﻣﻴﻠﻲ 6 ﮔـﺮم در دﺳـﻲﻟﻴﺘﺮ ﺑﻪ ﻋﻨﻮان ﻣﺤﺪوده اﺳـﺘﺎﻧﺪارد ﺑـﺮای ﻣﺮاﻗﺒـﺖ از ﻣﺒﺘﻼﻳـﺎن ﻓﻨﻴـﻞ ﻛﺘﻮﻧﻮری در ﻧﻈﺮ ﮔﺮﻓﺘﻪ ﺷﺪ [13]. اﮔﺮ ﭼﻪ ﭘﺎﺗﻮژﻧﺰ آﺳﻴﺐ ﻣﻐﺰی در ﻛﺘﻮﻧﻮری ﻓﻨﻴﻞ ﻛﺎﻣﻼ ﺷﻨﺎﺧﺘﻪ ﻧـﺸﺪه، اﻣﺎ ﺑﺪﻳﻬﻲ اﺳﺖ ﻛﻪ اﻳﻦ وﺿﻌﻴﺖ ﺑﺎ اﻓﺰاﻳﺶ ﻣﺰﻣﻦ ﻓﻨﻴﻞ آﻻﻧـﻴﻦ ﺧـﻮن ارﺗﺒﺎط دارد [14] ﺑﻄﻮرﻳﻜﻪ وﻗﺘﻲ در ﻃﻲ دوره ﺣﻴﺎﺗﻲ ﺗﻜﺎﻣﻞ ﻣﻐﺰ ﻣﻘـﺪار ﻓﻨﻴﻞ آﻻﻧـﻴﻦ ﺑـﺎﻻﺗﺮ از ﺣـﺪ ﻧﺮﻣـﺎل ﺑﺎﺷـﺪ، ﺗﺠﻤـﻊ ﻓﻨﻴـﻞ آﻻﻧـﻴﻦ ﻳـﺎ ﻣﺤﺼﻮﻻت ﻛﺎﺗﺎﺑﻮﻟﻴﻚ آن، ﻛﻤﺒﻮد ﺗﻴﺮوزﻳﻦ ﻳﺎ ﻣﺤﺼﻮﻻت آن و ﻳـﺎ ﻫـﺮ ﭼﻬﺎر وﺿﻌﻴﺖ ﺑﻄﻮر ﺗﻮام رخ دﻫﺪ، ﺳﺒﺐ ﺗﻮﻟﻴـﺪ ﺳـﻠﻮﻟﻬﺎی ﻋـﺼﺒﻲ ﺑـﺎ دﻧﺪرﻳﺖ ﻫﺎ و آﻛﺴﻮن ﻫﺎی ﺗﻐﻴﻴﺮ ﺷﻜﻞ ﻳﺎﻓﺘﻪ و ﻧﻘﺺ در ﺻﻔﺤﺎت ﻣﻴﻠﻴﻦ ﺷﺪه و از ﺑﻴﻦ رﻓﺘﻦ ﻣﻴﻠﻴﻦ ﺑﺎﻋﺚ ﻛﻮﺗـﺎه ﺷـﺪن ﻣـﺴﻴﺮ اﻳﻤﭙـﺎﻟﺲ ﻫـﺎی ﻋﺼﺒﻲ و ﻧﻘﺺ در ارﺗﺒﺎط ﺑﻴﻦ ﺳﻠﻮﻟﻬﺎی ﻋـﺼﺒﻲ ﻣـﻲ ﺷـﻮد . ﻫﻤﭽﻨـﻴﻦ ﺑﻌﻠﺖ ﻣﻬﺎر رﻗﺎﺑﺘﻲ در اﺛﺮ ﻏﻠﻈﺖ ﺑﺎﻻی ﻓﻨﻴﻞ آﻻﻧﻴﻦ روی ﺟﺬب ﺳـﺎﻳﺮ آﻣﻴﻨﻮاﺳﻴﺪﻫﺎی ﻃﺒﻴﻌﻲ ﺑﺰرگ ﺑﻪوﻳﮋه ﻣﺘﻴﻮﻧﻴﻦ ﺗﻴﺮوزﻳﻦ و از ﺳﺪ ﺑـﻴﻦ ﺧﻮن و ﻣﻐﺰ، ﺗﻮﻟﻴﺪ ﻧﻮروﺗﺮاﻧﺴﻤﻴﺘﺮﻫﺎ ﺑـﻮﻳﮋه ﺳـﺮوﺗﻮﻧﻴﻦ و دوﭘـﺎﻣﻴﻦ در ﻣﻐﺰ ﻛﺎﻫﺶ ﻳﺎﻓﺘﻪ و ﻣﻘﺪار ﭘﺎﺋﻴﻦ دوﭘﺎﻣﻴﻦ در ﻣﻐﺰ ارﺗﺒﺎط ﻃﺒﻴﻌﻲ ﺑـﻴﻦ ﺳﻠﻮﻟﻬﺎی ﻋـﺼﺒﻲ را ﻣﺨﺘـﻞ ﻣـﻲ ﻛﻨـﺪ ﻛـﻪ ﺳـﺒﺐ ﻧﻘـﺺ در ﻋﻤﻠﻜـﺮد ﺷﻨﺎﺧﺘﻲ ﻣـﻲ ﮔـﺮدد و ﻋـﺪم ﺗﻌـﺎدل در ﻏﻠﻈـﺖ اﺳـﻴﺪﻫﺎی آﻣﻴﻨـﻪ در ﺳﻴﺴﺘﻢ ﻋﺼﺒﻲ ﺳﺒﺐ اﻳﺠﺎد ﻣﺸﻜﻼت رﻓﺘﺎری و ﻃﻮﻻﻧﻲ ﺷـﺪن زﻣـﺎن ﻋﻤﻠﻜﺮد ﻣﻲﺷﻮد [18-14].
-

-
اﺧﺘﻼﻻت ﻣﺘﺎﺑﻮﻟﻴﻜﻲ وﻳﮋﮔﻲ ﻫﺎی ژﻧﺘﻴﻜﻲ ﻫﺴﺘﻨﺪ ﻛﻪ ﺑﻪ ﻋﻠﺖ ﻧﺒـﻮد ﻳـﺎ ﻛــﺎﻫﺶ ﻓﻌﺎﻟﻴــﺖ ﻳــﻚ آﻧــﺰﻳﻢ ﻳــﺎ ﻛﻮﻓــﺎﻛﺘﻮر اﺧﺘــﺼﺎﺻﻲ ﺑﻮﺟــﻮد ﻣﻲآﻳﻨﺪ [20،19،13]ﻛﻪ در آﻧﻬﺎ ﻣﻌﻤﻮﻻ ﻣﺴﻴﺮ ﻣﺘﺎﺑﻮﻟﻴﺴﻢ ﻳﻚ اﺳﻴﺪ آﻣﻴﻨـﻪ درﮔﻴﺮ ﻣﻲﺑﺎﺷﺪ ﻛﻪ ﺳﺒﺐ ﺗﺠﻤﻊ ﻣﻘﺎدﻳﺮ ﺳـﻤﻲ اﻳـﻦ اﺳـﻴﺪ آﻣﻴﻨـﻪ در ارﮔﺎن ﻣﺜﻞ ﻫﺎﻳﻲﻛﺒﺪ، ﭘﻮﺳـﺖ، ﭼـﺸﻢ و ﻣﻐـﺰ ﻣـﻲ ﺷـﻮد. درمان نیز ﻣﺴﺘﻘﻴﻤﺎ ﻣﺘﺎﺑﻮﻟﻴﺴﻢ ﻣﺴﻴﺮ ﺧﺎص اﺳﻴﺪ آﻣﻴﻨﻪ را ﺑﺎ اﺳـﺘﻔﺎده از رژﻳـﻢ ﻏﺬاﻳﻲ ﺗﺤﺖ ﺗﺎﺛﻴﺮ ﻗﺮار ﻣﻲدﻫﺪ [21]. از ﺟﻤﻠﻪ اﻳﻦ ﺑﻴﻤﺎری ﻫﺎ ﻫﻴﭙﺮﻓﻨﻴﻞآﻻﻧﻴﻨﻤﻴﺎ ﻣﻲﺑﺎﺷﺪ ﻛﻪ در آن ﻣﺘﺎﺑﻮﻟﻴـﺴﻢ اﺳـﻴﺪ آﻣﻴﻨـﻪ ﻓﻨﻴـﻞ آﻻﻧـﻴﻦ ﻣﺴﻴﺮ ﻃﺒﻴﻌﻲ ﺧﻮد را ﻃﻲ ﻧﻤﻲ ﻛﻨﺪ. اﻳﻦ ﺑﻴﻤﺎری ﭼﻬـﺎر ﻧـﻮع ﻣﺨﺘﻠـﻒ دارد:[13]
-
1. ﻛﻼﺳﻴﻚ ﺷﻜﻞ ﻛﺘﻮﻧﻮری ﻓﻨﻴﻞ
ﺷﺎﻳﻊ ﺗﺮﻳﻦ ﻓﺮم ﻫﻴﭙﺮﻓﻨﻴﻞ آﻻﻧﻴﻨﻨﻤﻲ [13] و ﻳﻜﻲ از ﺷﺎﻳﻊ ﺗﺮﻳﻦ اﺧـﺘﻼﻻت ارﺛﻲ ﻣﺘﺎﺑﻮﻟﻴﻚ اﺳﺖ [22،14] ﻛﻪ ﺑﻪ ﻋﻠﺖ ﻓﻘﺪان ﻳﺎ ﻛﺎﻫﺶ ﻓﻌﺎﻟﻴﺖ آﻧـﺰﻳﻢ ﻓﻨﻴﻞ آﻻﻧﻴﻦ ﻫﻴﺪروﻛﺴﻴﻼز در ﻛﺒـﺪ رخ ﻣـﻲ دﻫـﺪ ﻛـﻪ اﻳـﻦ ﻧﻘـﺺ از ﺗﺒﺪﻳﻞ ﻓﻨﻴﻞ آﻻﻧﻴﻦ ﺑﻪ ﺗﻴﺮوزﻳﻦ ﻣﻤﺎﻧﻌﺖ ﻣﻲ ﻛﻨﺪ [21].
ﻋﻼﺋـﻢ ﺑﻴﻮﺷﻴﻤﻴﺎﻳﻲ ﺑﻪ ﺻﻮرت ﻏﻠﻈﺖ ﺧﻮﻧﻲ ﻓﻨﻴﻞ آﻻﻧﻴﻦ ﺑﺎﻻﺗﺮ از 20 ﻣﻴﻠـﻲﮔﺮم در دﺳﻲ ﻟﻴﺘﺮ و اﻓﺰاﻳﺶ ﻓﻨﻴﻞ ﻛﺘﻮن ﻫﺎ در ادرار [13] و ﻋﻼﺋـﻢ ﺑـﺎﻟﻴﻨﻲ ﺷﺎﻣﻞ ﻣﻮی روﺷﻦ و ﭼﺸﻢ آﺑﻲ، اﮔﺰﻣﺎ، اﺣﺴﺎس ﺑﻮی ﻛﭙـﻚ در ادرار و ﻋـــﺮق [21]، ﺗـــﺎﺧﻴﺮ ﺗﻜﺎﻣـــﻞ، ﻣﻴﻜﺮوﺳـــﻔﺎﻟﻲ [16 ،15]، ﻧﺎﻫﻨﺠـــﺎری ﻫـــﺎی اﻟﻜﺘﺮواﻧﺴﻔﺎﻟﻮﮔﺮام،ﭘﺮ ﻓﻌﺎﻟﻴﺘﻲ [17]، ﺣﺮﻛﺖﻫـﺎی ﻏﻴـﺮ ﻃﺒﻴﻌـﻲ، ﻟـﺮزش، ﺗﺎﺧﻴﺮ درﻛﺴﺐ ﻣﻬﺎرﺗﻬﺎی اﺟﺘﻤﺎﻋﻲ [23]، ﺗﺎﺧﻴﺮ در ﺗﻜﻠـﻢ [16] و ﺻـﺮع اﺳﺖ [24]. درﻣﺎن ﺷﺎﻣﻞ ﻛﺎﻫﺶ ﻓﻨﻴـﻞ آﻻﻧـﻴﻦ و اﻓـﺰاﻳﺶ ﺗﻴـﺮوزﻳﻦ رژﻳـﻢ ﻏﺬاﻳﻲ و ﺣﻔﻆ ﻓﻨﻴﻞ آﻻﻧﻴﻦ ﺳﺮم ﺑﻴﻦ 2 ﺗﺎ 6 ﻣﻴﻠﻲ ﮔﺮم در دﺳـﻲ ﻟﻴﺘـﺮ اﺳﺖ [13].
-
2. ﺷﻜﻞﺧﻔﻴﻒ ﻛﺘﻮﻧﻮری ﻓﻨﻴﻞ
ﺑﻪ ﻋﻠﺖ ﻧﻘﺺ آﻧﺰﻳﻢ ﻓﻨﻴﻞ آﻻﻧﻴﻦ ﻫﻴﺪروﻛﺴﻴﻼز اﻳﺠﺎد ﻣﻲ ﺷﻮد و ﻋﻼﺋﻢ ﺑﻴﻮﺷﻴﻤﻴﺎﻳﻲ ﺷﺎﻣﻞ ﻏﻠﻈﺖ ﺧﻮﻧﻲ ﻓﻨﻴﻞ آﻻﻧﻴﻦﺑﻴﺸﺘﺮ از 6 ﻣﻴﻠـﻲ ﮔـﺮم در دﺳﻲ ﻟﻴﺘـﺮ و اﻓـﺰاﻳﺶ ﻓﻨﻴـﻞ ﻛﺘﻮنﻫﺎ در ادرار اﺳﺖ. درﻣﺎن از ﻃﺮﻳﻖ ﻛﺎﻫﺶ ﻣﻘﺪار ﻓﻨﻴﻞ آﻻﻧـﻴﻦ و اﻓﺰاﻳﺶ ﻣﻘﺪار ﺗﻴﺮوزﻳﻦ در ﺣﻔﻆ رژﻳﻢ ﻏﺬاﻳﻲ و آﻻﻧﻴﻦ ﻓﻨﻴﻞ ﺳـﺮم در ﻣﺤﺪوده 2 ﺗﺎ 6 ﻣﻴﻠﻲ ﮔﺮم در دﺳﻲ ﻟﻴﺘﺮ اﻧﺠﺎم ﻣﻲ ﺷﻮد [13].
-
3. ﻓﻨﻴﻞ ﻛﺘﻮﻧﻮری ﺟﻨﻴﻨﻲ
در اﻳﻦ ﺣﺎﻟﺖ ﻫﻴﭽﮕﻮﻧﻪ ﻧﻘﺺ آﻧﺰﻳﻤﻲ ﻣﺸﺎﻫﺪه ﻧﻤﻲ ﺷـﻮد ﺑﻠﻜـﻪ وﻗﺘـﻲ رخ ﻣﻲ دﻫﺪﻛﻪ ﻏﻠﻈﺖ ﻓﻨﻴﻞ آﻻﻧﻴﻦ در ﺧﻮن زن ﺑـﺎردار ﺑـﺎﻻﺗﺮ از ﺣـﺪ ﻃﺒﻴﻌﻲ ﺑﺎﺷﺪزﻳﺮا ﻓﻨﻴﻞ آﻻﻧﻴﻦ ﺑـﻪ ﻃـﻮر ﻓﻌـﺎل از ﺟﻔـﺖ ﺑـﻪ ﺟﻨـﻴﻦ ﻣﻨﺘﻘﻞ و ﻏﻠﻈﺖ ﭘﻼﺳﻤﺎﻳﻲ ﻓﻨﻴـﻞ آﻻﻧـﻴﻦ ﺟﻨـﻴﻦ 1، 1/5 ﺗـﺎ 2 ﺑﺮاﺑـﺮ ﺧﻮن ﻣﺎدر ﺷﺪه و اﻳﻦ اﻓﺰاﻳﺶ ﺑﻪ وﺳﻴﻠﻪ ﺳﺪ ﺑﻴﻦ ﺧﻮن و ﻣﻐـﺰ ﺟﻨـﻴﻦ ﻣﺠﺪدا 2 ﺗﺎ 4 ﺑﺮاﺑﺮ ﻣﻲ ﺷﻮد و در ﺻـﻮرﺗﻲ ﻛـﻪ ﻏﻠﻈـﺖ ﻓﻨﻴـﻞ آﻻﻧـﻴﻦ ﺳﻴﺴﺘﻢ ﻋﺼﺒﻲ ﺑﻪ ﺑﺎﻻﺗﺮ از 600 ﻣﻴﻜﺮوﻣﻮل در ﻟﻴﺘﺮ ﺑﺮﺳـﺪ ﺑـﺎ ﺗﻜﺎﻣـﻞ ﻣﻐﺰ ﺗﺪاﺧﻞ ﺧﻮاﻫﺪ داﺷﺖ. ﻋﻼﺋﻢ ﻛﻠﻴﻨﻴﻜﻲ ﺑﻪﺻـﻮرت آﺳـﻴﺐ ﻣﻐـﺰی ﺟﻨﻴﻦ و اﻓﺰاﻳﺶ ﻣﻴـﺰان وﻗـﻮع ﻧﻘـﺎﺋﺺ ﻗﻠﺒـﻲ، ﻋﻘـﺐ اﻓﺘـﺎدﮔﻲ رﺷـﺪ ﺟﺴﻤﻲ و ذﻫﻨﻲ و ﻣﻴﻜﺮوﺳﻔﺎﻟﻲ اﺳﺖ و ﻫﻴﭽﮕﻮﻧﻪ درﻣﺎن رژﻳﻤﻲ ﻧﺪارد. ﺗﻨﻬﺎ راه ﭘﻴﺸﮕﻴﺮی ﻛﻨﺘﺮل ﺟﺪی ﻣﻴـﺰان ﻓﻨﻴـﻞ آﻻﻧـﻴﻦ ﻣـﺎدر ﻗﺒـﻞ و درﺗﻤﺎم دوره ﺑﺎرداری اﺳﺖ [13].
-
4. ﻓﻨﻴﻞ ﻛﺘﻮﻧﻮریﺑﺪﺧﻴﻢ [14]
ﺑﻪ دﻟﻴﻞ ﻧﻘـﺺ در ﺳـﻨﺘﺰ ﻳـﺎ ﺗـﺸﻜﻴﻞ ﻣﺠـﺪد ﻛﻮﻓـﺎﻛﺘﻮر آﻧـﺰﻳﻢ دی ﻫﻴﺪروﭘﺘﺮﻳﺪﻳﻦ ردوﻛﺘﺎز ﻳﻌﻨـﻲ ﺗﺘﺮاﻫﻴـﺪروﺑﻴﻮﭘﺘﺮﻳﻦ اﻳﺠـﺎد ﻣـﻲ ﺷـﻮد. ﻋﻼﻣﺖ ﺑﻴﻮﺷﻴﻤﻴﺎﻳﻲ ﺷﺎﻣﻞ ﻏﻠﻈﺖ ﺧـﻮﻧﻲ ﻓﻨﻴـﻞ آﻻﻧـﻴﻦ ﻛﻤﺘـﺮ از 20 ﻣﻴﻠﻲ ﮔﺮم دردﺳﻲﻟﻴﺘﺮ و ﻋﻼﺋـﻢﺑـﺎﻟﻴﻨﻲ ﺗﺤﺮﻳـﻚ ﭘـﺬﻳﺮی، ﺗـﺎﺧﻴﺮ در ﺗﻜﺎﻣﻞ و ﺗﺸﻨﺞ اﺳﺖ [13].
-
فنیل آلانین و نقش آن در بدن
فنیل آلانین (مواد گوشتی (پروتئین ها)، لبنیات و حبوبات ) یک اسید آمینه ضروریست که از طریق مواد غذائی وارد بدن میشود. تنها قسمت کوچکی از فنیل آلانین موجود در بدن برای ساخت پروتئینها بکار میرود.بقیه از طریق هیدروکسیلاسیون آنزیماتیک تبدیل به تیروزین میشود.تیروزین پیشساز هورمونهای تیروئیدی، کاتکل آمینها و ملاتونین است. فنیل آلانین که به طور خلاصه «فی» خوانده می شود، جزو مواد ضروری در سلامت انسان است. این ماده در ترکیب پروتئین ها موجود است و با غذا وارد بدن می شود.در نوزادان مبتلا به این بیماری مصرف غذاهای پروتئینی از جمله شیر مادر و یا شیر خشک معمولی باعث افزایش شدید غلظت خونی «فی» و تجمع آن در بافت های مختلف بدن می شود و رشد و تکامل مغز و اعصاب را مختل می کند و به ضایعه مغزی و یا عقب ماندگی ذهنی پایدار منجر می شود. هیدروکسیلاسیون فنیل آلا نین بوسیله آنزیم فنیل آلانین هیدروکسیلاز (PAH) که در کبد ساخته میشود صورت میگیرد.
برای انجام این واکنش حضور تتراهیدروبیوپترین(BH4) ضروریست. اگر فنیل آلانین بدلیل نقائص آنزیمی تبدیل به تیروزین نشود از طریق مسیر دیگری تبدیل به متابولیتهای دیگری مثل فنیل پیرویک اسید شده و سرانجام از کلیه ها دفع خواهد شد. اکثر افراد مبتلا به هیپر فنیل آلانینمی (99%-90) دچار نقص در آنزیمPAH هستند. این نقص آنزیمی بصورت اتوزوم مغلوب منتقل شده ودر 1:10000 افراد دیده میشود. طبق آمار موجود تعداد معلولین "پی ک یو" در کشور، به بیش از 6000 نفر رسیده که نیمی از آنها هنوز ناشناخته مانده اند. در صورت وجود فعالیت خفیف این آنزیم افزایش سطح فنیل آلانین در گردش خون مشاهده میشوداما سطح متابولیتهای آنها به حدی نیست که در ادرار ترشح شود(Non-PKU Hyperphenylalaninemia). در شکل کلاسیک PKU فعالیت PAH وجود ندارد و غلظت فنیل پیرویک اسید در ادرار بالا است. migna.ir در موارد نادر شیرخواران مبتلا به نوع خاصی از فنیل کتونوری بنام PKU بدخیم هستند که ناشی از نقص در سنتز و متابولیسم BH4 می باشد.
-
بیماریزایی
فنیل آلانین پس از ورود به بدن توسط آنزیمی به نام فنیل آلانین هیدروکسیلاز شکسته و به تیروزین تبدیل میشود. کوآنزیم این واکنش، تتراهیدروبیوپترن (BH4) میباشد. سپس تیروزین شکسته شده و به مواد متعددی از جمله رنگ دانه پوست و مو تبدیل شده و متابولیتهای نهایی آن از بدن دفع میشود. پس چنانچه آنزیم «فنیل آلانین هیدروکسیلاز» که فقط در کبد ساخته میشود به دلیل اختلالات ژنی وجود نداشته باشد «فنیل آلانین» وارد شده به بدن، در بافتهای مختلف از جمله مغز تجمع یافته و سبب آسیبهای متعددی به بافت مغز میشود.
-
تنها راه سنتز تیروزین در انسان از همین طریق است. در افراد مبتلا به فنیل کتونوری تیروزین، اسید آمینه ضروری میشود و باید از غذا تامین شود. در این افراد فنیل آلانین خون نیز به میزان زیادی بالا میرود. در افراد طبیعی مقدار کمی از فنیل آلانین، به فنیل پیروات، فنیل استات و فنیل لاکتات تبدیل میشود اما در افراد مبتلا به فنیل کتونوری به علت بالا بودن میزان فنیل آلانین، مقدار زیادی از این فنیل کتونها تولید شده که وارد خون و ادرار میشوند.
مقدار زیاد فنیل پیروات در خون، از عمل آنزیم پیروات دکربوکسیلاز در مغز بصورت آلوستریک، جلوگیری میکند. این عمل به نوبه خود در تشکیل میلین اشکال ایجاد میکند و منجر به عقب ماندگی ذهنی میشود. یکی دیگر از دلایل عقب ماندگی ذهنی در افراد مبتلا به فنیل کتونوری کاهش تولید نوروترانسمیترهایی مانند دوپامین میباشد.
تیروزین پیش ساز سنتز این نوروترانسمیترها است. در این بیماران مقدار دوپامین و سروتونین در ادرار کمتر از حد طبیعی است. مصرف غذاهای پروتئینی از جمله شیرخشکهای معمولی و به میزان کمتر شیرمادر باعث افزایش شدید غلظت خونی فنیل آلانین و تجمع آن در بدن، اختلال در تکامل مغز و اعصاب و در نهایت ضایعه مغزی و عقب ماندگی ذهنی پایدار در مبتلایان میشود.
تجمع فنیل آلانین و مشتقات غیرطبیعی آن در نسوج مختلف، فعالیت سلولها، به خصوص سلولهای سیستم عصبی را تحت تأثیر قرار داده و از رشد آن جلوگیری میکند. این ضایعات متأسفانه در هفتههای اول تولد، نشانههای واضحی ندارند در نتیجه بیماری بموقع تشخیص داده نشده و درمان آن به تأخیر میافتد.
در چنین دوران بحرانی که حیاتیترین مرحله شکلگیری و تکامل مغز کودک است، تغذیه با شیرمادر یا شیرخشک معمولی و یا هر ماده پروتئینی دیگر اگر ادامه یابد منجر به ضایعه عصبی میشود و توان هوشی و قدرت ادراک کودک به تدریج تضعیف میشود و در نهایت منجر به معلولیت شدید ذهنی میگردد. متأسفانه گذشت زمان، شانس طلایی درمان را از بین برده و رژیم غذایی و درمانهای بعدی، دیگر نتیجه مطلوب را نخواهند داشت.
-
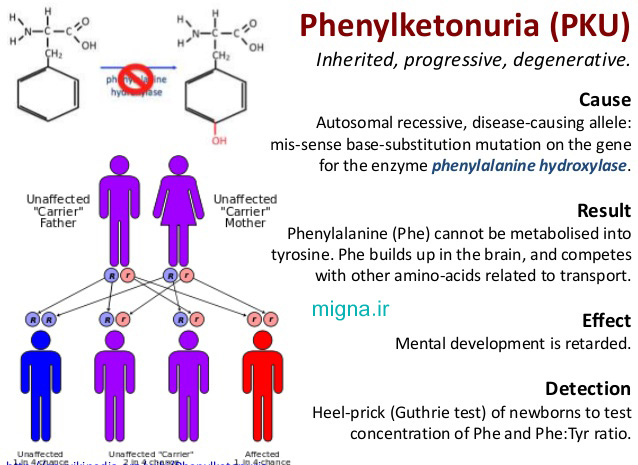
این بیماری مانند تالاسمی به صورت اتوزوم مغلوب (aa) به ارث میرسد. و این یعنی جنسیت فرد در ابتلا به آن نقش مستقیم و ویژه ای ندارد،ژن این بیماری بر روی کروموزم ۱۲ قرار گرفتهاست. چنانچه والدین هردو حامل این ژن باشند و خود سالم باشند(که معمولاً در ازدواجهای خویشاوندی این احتمال بالاتر است) در تولد هر فرزندانشان ۲۵درصد احتمال ابتلا به فنیل کتونوریا وجود دارد. البته از آن جا آزمایش تشخیص این بیماری بسیار آسان و کم هزینه است،در بیمارستان ها و زایشگاه های بسیاری از کشور های پیشرفته،همه نوزادان را از نظر دارا بودن عامل این بیماری آزمون میکنند. به طور کلی توصیه میشود افراد قبل از هرگونه اقدام در زمینه ازدواج به مشاوران ژنتیک حتماً مراجعه بکنند به ویژه افرادی که در خانواده آنان سابقه ابتلا به هریک از بیماری های ژنتیکی وجود دارد.
-
علائم و نشانه های بیماری
کودک مبتلا به بیماری «فنیل کتونوری» در ابتدای تولد بدون علامت است. اما به تدریج در پایان ماههای اول دچار تأخیر در تکامل، استفراغ، کاهش رشد، روشن شدن رنگ موهای سر و چشم و تشنج میشود. سپس با افزایش سن، کوچکی دورسر، بیقراری، کاهش توجه، حرکات تکراری دستها و اندامها و عقبماندگی ذهنی بروز میکند. همچنین ادرار و تنفس این کودکان به دلیل وجود فراوردههای فنیل آلانین، بوی کپک میدهد و ممکن است راشهای پوستی (کهیر) در بدن کودکان مبتلا مشاهده شود که با رشد کودک از بین میرود. این بیماری در بدو تولد هیچ گونه نشانه بارزی ندارد و نوزاد در ۲ تا ۳ ماه اول زندگی ظاهر کاملا سالمی دارد ولی به تدریج علایمی هم چون بی میلی به خوردن شیر، استفراغ بعد از خوردن شیر، بروز اگزما و جوش در سطح بدن، بورشدن موهای سر بدون سابقه ارثی ظاهر می شود.عرق بدن و ادرار این نوزادان اغلب بوی زننده و بسیار نامطبوع کپک مانند دارد. با گذشت زمان کودک دچار عقب ماندگی ذهنی می شود. این کودکان اغلب ناآرام و پرجنب و جوش اند و تعادل عصبی خوبی ندارند.قدرت تکلم این کودکان ضعیف و راه رفتن آن ها دچار مشکل می شود که ممکن است برای همیشه باقی بماند.
-
تشخیص
تظاهرات بالینی بیمار تا ۶- ۵ ماهگی بسیار گمراه کننده است. متاسفانه تشخیص اغلب ، زمانی اتفاق می افتد که بیماری منجر به عقب ماندگی ذهنی کودک شده است و ضایعه مغزی به وجود آمده درمان ناپذیر شده است.
بنابراین تنها و بهترین راه تشخیص این بیماری اندازه گیری غلظت خونی «فی» در بدو تولد نوزاد است. اینآزمایش باید هرچه زودتر در روزهای اول تولد انجام شود، زیرا تاخیر در تشخیص بیماری از هفته سوم به بعد خطرناک است و ممکن است صدمات مغزی به معلولیت دائم منتهی شود. این بیماری از حدود ۴۰ سال پیش شناخته شده و امروزه یک بیماری کنترل شده است.
در کشورهای پیشرفته در بدو تولد با چند قطره خون که از پاشنه پای کودک گرفته میشود، میتوانند در حدود ۳۰ بیماری ژنتیکی از جمله پی کی یو را تشخیص دهند که اکثر این بیماریها قابل درمان هستند[25].
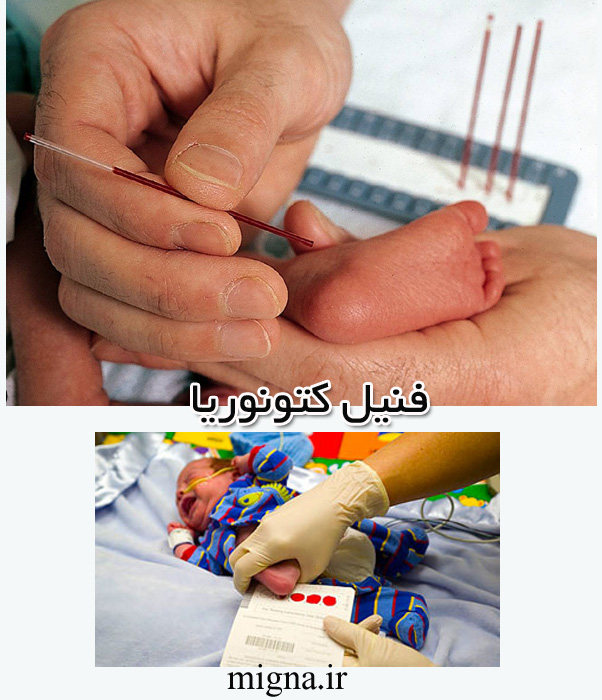
برای اندازهگیری سطح «فنیل آلانین» خون باید ۴۸ تا ۷۲ ساعت بعد از تولد کودک تست انجام پذیرد. اگر سطح «فنیل آلانین» بالاتر از ۲۰ میلیگرم در دسیلیتر باشد و سطح تیروزین نرمال و متابولیتهای ادراری «فنیل آلانین» بالا باشد و از طرفی غلظت فاکتور تتراهیدروبیوپترین هم نرمال باشد، تشخیص «فنیل کتونوریا» مسجل است.
خوشبختانه با شناخت ژن این بیماری امروزه تشخیص آن در ماههای اول حاملگی نیز امکانپذیر است. در حال حاضر با آزمایش ژنتیک بیمار و والدین نقص ژنتیکی تشخیص داده میشود و بررسی جنین در حاملگیهای بعدی مادر ممکن است.
در صورت تشخیص ابتلای جنین به بیماری، امکان سقط وی وجود دارد. همچنین با مشخص شدن نقص ژنتیکی، امکان مشاوره افراد فامیل بیمار که قصد دارند با یکدیگر ازدواج کنند، وجود خواهد داشت. اگر یک زوج ناقل بیماری باشند، براساس اطلاعات کسب شده در مشاوره ژنتیک، میتوانند برای زندگی آینده خود تصمیم بگیرند.
باید در نظر داشت که مثبت بودن نتیجه آزمایش همیشه دلیل بر ابتلای نوزاد به بیماری نیست، زیرا ممکن است کبد بعضی از نوزادان به خصوص آن هایی که نارس به دنیا آمده اند، در روزهای اول تولد هنوز به طور کامل فعال نشده باشد، در نتیجه«فی» خون آن ها به طور طبیعی افزایش نشان می دهد.ولی چند روز بعد با رشد نوزاد، فعالیت کبد به وضع طبیعی بر می گردد و نتیجه آزمایش اصلاح می شود بنابراین در این شرایط تمام آزمایش ها بعد از ۷ تا ۸ روز باید دوباره تکرار شود تا نتیجه اول تایید شود.در این مدت و تا مشخص شدن نتیجه نهایی نباید نوزاد را از شیر مادر یا غذای متعارف محروم کرد. جواب طبیعی در مرحله اول و یا دوم نشانه سلامت نوزاد است. migna.ir ولی نتیجه مثبت در هر ۲ نوبت دلیل بر این است که نوزاد باید تا مدتی تحت نظر متخصص کودکان و کارشناس تغذیه قرار گیرد و با رژیم غذایی مخصوص مداوا شود[25].
-
زمان درمان و درمان
در صورت تشخیص زودرس و شروع شیر مخصوص «فنیل کتونوری» از ابتدای نوزادی پیشآگهی خوب خواهد بود و کودک مبتلا میتواند از هوش خوب و رفتار مناسب برخوردار شود. ولی تأخیر در درمان به بروز عقبماندگی ذهنی، کوچکی دورسر و اختلالات رفتاری منجر خواهد شد.
هدف از درمان این بیماری، کاهش مقدار فنیل آلانین در بدن به منظور پیشگیری از عقب ماندگی ذهنی کودک است. این بیماری در اکثرِ موارد از توارث پذیری بسیار بالایی برخوردار است و از سوی دیگر این بیماری درمان دارویی ندارد.ولی از طریق رژیم مناسب غذایی میتوان سطح «فنیل آلانین» را در حد نرمال نگه داشت و از این رو با تشخیص زودرس بیماری (از روز سوم تولد) و شروع تغذیه کودک با شیرهای مخصوص سطح سرمی «فنیل آلانین» در ۱۲ سال اول زندگی بین ۲ تا ۶ میلیگرم در دسیلیتر و بعد از آن تا ۲ تا ۱۰ میلیگرم در دسیلیتر نگه داشته میشود[25]. ولی باید به موازات آن ویتامینها، کلسیم و کالری کافی به کودک برسد. بعد از ۶ ماهگی نیز غذاهای مخصوص بتدریج شروع میشود.
-
تغذیه
با افزایش سن چون کودک به تغذیه نیاز بیشتری دارد میتواند از سیبزمینی، سبزیها، انواع میوه، نشاسته، چربی، برنج و نانها و به مقدار کمتر از حبوبات و شیرمخصوص استفاده نماید. البته در فواصل منظم باید سطح «فنیل آلانین» سرم اندازهگیری شود. رژیم غذایی هر بیمار با توجه به سطح فنیل آلانین بایستی توسط کارشناس تغذیه تعیین گردد. همچنین عدم ادامه رژیم غذایی مناسب در بزرگسالی منجر به اشکال در بهره هوشی و عملکرد شناختی بیمار میشود بنابراین توصیه میشود بیماران رژیم محدود از فنیل آلانین را برای همه عمر رعایت کنند.
-
خلاصه و نتیجه گیری:
ﻓﻨﻴﻞ ﻛﺘﻮﻧﻮری ﻳﻚ ﺧﻄﺎی ﻣﺘﺎﺑﻮﻟﻴﺴﻢ اﺳﺖ ﻛﻪ ﺑﻪ ﻋﻠﺖ ﻧﻘﺺ در آﻧﺰﻳﻢ ﻓﻨﻴﻞ اﻻﻧﻴﻦ ﻫﻴﺪروﻛﺴﻴﻼز ﻛﻪ ﻓﻨﻴﻞ اﻻﻧﻴﻦ را ﺑﻪ ﺗﻴﺮوزﻳﻦ ﺗﺒﺪﻳﻞ ﻣﻲ ﻛﻨﺪ اﻳﺠﺎد ﻣﻲ ﺷﻮد. اختلال اصلی در این بیماری، تجمع اسید آمینه فنیل آلانین در مایعات بدن و سیستم عصبی است. این بیماری از طریق ژنهای مغلوب وکروموزوم های معمولی از والدین به کودک منتقل می شود و موجب بروزعقب ماندگی نسبتاً شدیدمی گردد. ﺑ
ﺪون درﻣﺎن اﻏﻠﺐ ﺑﻴﻤﺎران دﭼﺎر ﻋﻘﺐ اﻓﺘﺎدﮔﻲ ذﻫﻨﻲ ﻣﻲ ﺷﻮﻧﺪ. ﺑﻪ ﺧﻮﺑﻲ ﻣﺸﺨﺺ ﺷﺪه اﺳﺖ ﻛﻪ درﻣﺎن ﻣﻨﺎﺳﺐ ﻧﻮزادان در ﺧﻼل دو ﻫﻔﺘﻪ اول زﻧﺪﮔﻲ ﭘﻴﺎﻣﺪﻫﺎی ﺑﻴﻤﺎری را ﺗﻐﻴﻴﺮ ﻣﻲ دﻫﺪ و ﻣﻨﺠﺮ ﺑﻪ رﺷﺪ و ﻧﻤﻮ ﻃﺒﻴﻌﻲ در ﻧﻮزاد ﻣﻲ ﺷﻮد. ﺗﺸﺨﻴﺺ ﺳﺮﻳﻊ، ﺑﻪ اﻳﻦ ﻋﻠﺖ در اوﻟﻴﻦ روزﻫﺎی زﻧﺪﮔﻲ اﻗﺪام اﺳﺎﺳﻲ اﺳﺖ. ﻏﺮﺑﺎﻟﮕﺮی اﻳﻦ ﺑﻴﻤﺎری ﺗﻮﺳﻂ آزﻣﻮﻧﻬﺎی ﻏﺮﺑﺎﻟﮕﺮی ﻧﻮزادی ﻳﻚ اﻗﺪام ﺳﻼﻣﺖ اﺟﺘﻤﺎﻋﻲ اﺳﺖ ﻛﻪ در ﭘﻨﺠﺎه ﺳﺎل اﺧﻴﺮ اﻧﺠﺎم ﻣﻲ ﺷﻮد .
-
مریم بیشه (دانشجوی ارشد روان شناسی عمومی) واحد علوم و تحقیقات تهران - سایت میگنا
-
منابع و ماخذ:
References
1. Scriver CR, Kaufman S. Hyperphenylalaninemia: Phenylalanine hydroxylase deficiency. In: Scriver CR, Beaudet AL, Valle D, eds. The metabolic and molecular basis of inherited disea se .8th ed. New York, NY: McGraw-Hill Inc; 2001:1667-1724.
-
2. Acosta PB, Michals Matalon K. Nutrition management of patients with inherited disorders of aromatic amino acid metabolism . In: Acosta PB, ed. Nutrition managem ent of patients with inherited metabo lic disorders. 1st ed . Sudbury, MA: Jones and Bartlett Publishers; 2010:119-152.
-
3. Anastasoaie V, Kurzius L, Forbes P, Waisbren S. Stability of blood phenylalanine levels and IQ in children with phenylketonuria. Mol Genet Metab 2008;95(1-2):17–20.
-
4. Blau N, van Spronsen JM, Levy HL. Phenylketonuria. Lancet 2010 Oct 23;376(9750):1417-1427.
-
5. Van Spronsen FJ, Hoeksma M, Reijngoud DJ. Brain dysfunction in phenylketonuria: is phenylalanine the only possible cause? J Inherit Metab Dis 2009;32(1):46-51.
-
6. Guthrie R, Susi A. A simple phenylalanine method for detecting phenylketonuria in large populations of newborn infants. Pediatrics 1963;32:338-343.
-
7. Golbahar J, Honardar Z (2002). Selective Screening of Phenylketonuria, Tyrosinemia and Maple Syrup Urine Disease in Southern Iran. Iran J Med Sci. 27: 134-135.
-
8. Koochmeshgi J, Bagheri A, Hosseini-Mazinani SM. Incidence of phenylketonuria in Iran estimated from consanguineous marriages. J Inherit Metab Dis 2002; 25(1):80-81.
-
9. Senemar S, Ganjekarimi H, Fathzadeh M, Tarami B ,Barzgar M. Epidemiological and clinical study of phenylketonouria (PKU) disease in the national screening program of neonates ,Fars Province, Southern Iran. Iranian J Publ Health 2009 ;38(2):58-64.
-
10. Habib A, Fallahzadeh MH, Kazeroni HR, Ganjkarimi AH. Incidence of Phenylketonuria in Southern Iran. Iran J Med Sci 2010; 35:137-139.
-
11. Karamifar H, Ordoei M, Karamizadeh Z, Amirhakimi GH. Incidence of Neonatal Hyperphenylalaninemia in Fars Province, South Iran. Iran J Pediatr 2010; 20(2):216-220.
-
12. Vallian S, Barahimi E, Moeini H. Phenylketonuria in Iranian population: a study in institutions for mentally retarded in Isfahan. Mutat Res 2003 May 15; 526(1-2):45-52.
-
13. Cristine M, Trahms. Medical nutrition therapy for metabolic Disorders. In: Mahan KL, EscoottStump S (eds). Krause’s Food Nutrition and Diet Therapy. 12th ed. Philadelphia; Suanders. 2008; Pp: 1141-69.
-
14. Hendrisksz CJ, Walter JH. Update on phenylketonuria. Current Pediatr. 2004;14(5):400-6.
-
15. Elsas LJ, Acosta PhB. Inherited metabolic disease; Amino Acids, Organic Acids, and Galactose. In: Shils M, Shike M, Ross A, etal (eds). Modern Nutrition In Health and Disease. 10th ed. Baltimore; Lippincott Williams & Wilkins. 2006; Pp: 909-45.
-
16. National Institutes of Health Consensus Development Panel. National Institutes of Health Consensus Development Conference Statement Phenylketonuria: Screening and Management, October 16-18, 2000. Pediatrics. 2001; 108(4);972-82.
-
17. Surtees R, Blau N. The Neurochemistry of Phenylketonuria. Eur J Pediatr. 2000;159(Suppl 2); 109-13.
-
18. Ormazabal A, Artuch R, Vilaseca M. A, et al. Pathogenetic mechanisms in Phenylketonuria: disorders affecting the metabolism of neurotransmitters and the antioxidant system. Revista de Neurologia. 2004; 39(10);956-61.
-
19. Farhud D, Shalileh M. Diet therapy in Maple Syrup Urine Disease. Tehran; Special Education Organization. In press. (Persian)
-
20. Farhud D, Shalileh M. Diet therapy in Galactosemia. Tehran; Special Education Organization. In press. (Persian)
-
21. Seashore M. Amino acid disorders, Metabolic disorders. In: Kligman RM, Marcdante KJ, Jenson HB, Behrman RE (eds). Nelson Essential of Pediatrics. 5th ed. Philadelphia; Elsevier Saunders. 2006; Pp:243-69.
-
22. Farhud D, Shalileh M. Diet therapy in Phenylketonuria. Tehran; Special Education Organization. In press. (Persian)
-
23. Phenylketonuria. US National Library of Medicine. Available at: http://www.nlm.nih.gov/ medlineplus/ency/article/001166.htm. Access date: Aug 18, 2008.
-
24. Santos LL, Magalhaes Mde C, Januario JN, et al. The time has come: A new for PKU treatment. Genet Mol Res. 2006; 5(1):33-44.
1. Scriver CR, Kaufman S. Hyperphenylalaninemia: Phenylalanine hydroxylase deficiency. In: Scriver CR, Beaudet AL, Valle D, eds. The metabolic and molecular basis of inherited disea se .8th ed. New York, NY: McGraw-Hill Inc; 2001:1667-1724.
-
2. Acosta PB, Michals Matalon K. Nutrition management of patients with inherited disorders of aromatic amino acid metabolism . In: Acosta PB, ed. Nutrition managem ent of patients with inherited metabo lic disorders. 1st ed . Sudbury, MA: Jones and Bartlett Publishers; 2010:119-152.
-
3. Anastasoaie V, Kurzius L, Forbes P, Waisbren S. Stability of blood phenylalanine levels and IQ in children with phenylketonuria. Mol Genet Metab 2008;95(1-2):17–20.
-
4. Blau N, van Spronsen JM, Levy HL. Phenylketonuria. Lancet 2010 Oct 23;376(9750):1417-1427.
-
5. Van Spronsen FJ, Hoeksma M, Reijngoud DJ. Brain dysfunction in phenylketonuria: is phenylalanine the only possible cause? J Inherit Metab Dis 2009;32(1):46-51.
-
6. Guthrie R, Susi A. A simple phenylalanine method for detecting phenylketonuria in large populations of newborn infants. Pediatrics 1963;32:338-343.
-
7. Golbahar J, Honardar Z (2002). Selective Screening of Phenylketonuria, Tyrosinemia and Maple Syrup Urine Disease in Southern Iran. Iran J Med Sci. 27: 134-135.
-
8. Koochmeshgi J, Bagheri A, Hosseini-Mazinani SM. Incidence of phenylketonuria in Iran estimated from consanguineous marriages. J Inherit Metab Dis 2002; 25(1):80-81.
-
9. Senemar S, Ganjekarimi H, Fathzadeh M, Tarami B ,Barzgar M. Epidemiological and clinical study of phenylketonouria (PKU) disease in the national screening program of neonates ,Fars Province, Southern Iran. Iranian J Publ Health 2009 ;38(2):58-64.
-
10. Habib A, Fallahzadeh MH, Kazeroni HR, Ganjkarimi AH. Incidence of Phenylketonuria in Southern Iran. Iran J Med Sci 2010; 35:137-139.
-
11. Karamifar H, Ordoei M, Karamizadeh Z, Amirhakimi GH. Incidence of Neonatal Hyperphenylalaninemia in Fars Province, South Iran. Iran J Pediatr 2010; 20(2):216-220.
-
12. Vallian S, Barahimi E, Moeini H. Phenylketonuria in Iranian population: a study in institutions for mentally retarded in Isfahan. Mutat Res 2003 May 15; 526(1-2):45-52.
-
13. Cristine M, Trahms. Medical nutrition therapy for metabolic Disorders. In: Mahan KL, EscoottStump S (eds). Krause’s Food Nutrition and Diet Therapy. 12th ed. Philadelphia; Suanders. 2008; Pp: 1141-69.
-
14. Hendrisksz CJ, Walter JH. Update on phenylketonuria. Current Pediatr. 2004;14(5):400-6.
-
15. Elsas LJ, Acosta PhB. Inherited metabolic disease; Amino Acids, Organic Acids, and Galactose. In: Shils M, Shike M, Ross A, etal (eds). Modern Nutrition In Health and Disease. 10th ed. Baltimore; Lippincott Williams & Wilkins. 2006; Pp: 909-45.
-
16. National Institutes of Health Consensus Development Panel. National Institutes of Health Consensus Development Conference Statement Phenylketonuria: Screening and Management, October 16-18, 2000. Pediatrics. 2001; 108(4);972-82.
-
17. Surtees R, Blau N. The Neurochemistry of Phenylketonuria. Eur J Pediatr. 2000;159(Suppl 2); 109-13.
-
18. Ormazabal A, Artuch R, Vilaseca M. A, et al. Pathogenetic mechanisms in Phenylketonuria: disorders affecting the metabolism of neurotransmitters and the antioxidant system. Revista de Neurologia. 2004; 39(10);956-61.
-
19. Farhud D, Shalileh M. Diet therapy in Maple Syrup Urine Disease. Tehran; Special Education Organization. In press. (Persian)
-
20. Farhud D, Shalileh M. Diet therapy in Galactosemia. Tehran; Special Education Organization. In press. (Persian)
-
21. Seashore M. Amino acid disorders, Metabolic disorders. In: Kligman RM, Marcdante KJ, Jenson HB, Behrman RE (eds). Nelson Essential of Pediatrics. 5th ed. Philadelphia; Elsevier Saunders. 2006; Pp:243-69.
-
22. Farhud D, Shalileh M. Diet therapy in Phenylketonuria. Tehran; Special Education Organization. In press. (Persian)
-
23. Phenylketonuria. US National Library of Medicine. Available at: http://www.nlm.nih.gov/ medlineplus/ency/article/001166.htm. Access date: Aug 18, 2008.
-
24. Santos LL, Magalhaes Mde C, Januario JN, et al. The time has come: A new for PKU treatment. Genet Mol Res. 2006; 5(1):33-44.
-
25. آلبرت لنینگر، مایکل کاکس، دیویدلی نلسون،. اصول بیوشیمی لنینجر. ترجمه رضا محمدی. آییژ، ۱۳۸۵. ص.. شابک ۹۶۴-۸۳۹۷-۰۵-۸.
25. آلبرت لنینگر، مایکل کاکس، دیویدلی نلسون،. اصول بیوشیمی لنینجر. ترجمه رضا محمدی. آییژ، ۱۳۸۵. ص.. شابک ۹۶۴-۸۳۹۷-۰۵-۸.
